
Final Project Update
Aim: To assess whether senescent beta cells release EVs with beta cell-specific intracellular cargo
The rationale for this aim is that release of EVs is greatly increased in senescent cells as part of SASP and may play important roles in cell-to-cell communication both locally and systemically to contribute to immune regulation.
The objective of this aim is to characterize the EVs released from senescent cells for beta cell specific cargo and to investigate whether these EVs communicate to dendritic cells to alter antigen presentation.
Accomplishments
In the past year, we made significant progress in understanding and characterizing EVs from senescent beta cells.
Senescent β-cells export autoantigens via EVs in T1D. To determine whether senescent β-cells secrete putative autoantigens in EVs, we treated isolated islets from 14-week NOD mice with senolytic compounds ABT-737 or ABT-199, both of which trigger apoptosis selectively in senescent β-cells and isolated EVs from the conditioned media 48 h later, using a small-scale peptide pulldown approach (Fig. 1A). This approach of EV isolation was preferred due to the very small volumes of islet conditioned media. Western blot analysis for EV markers revealed a decrease in key sEV biogenesis markers Alix and Flotillin-1, while GAPDH and Actin were unchanged. Notably, β-cell autoantigens pro-insulin and IAPP were both decreased in islets treated with senolytic compounds as compared to controls, but GAD65 was unaffected (Fig. 1B). To validate these results, we carried out mass spectrometry on the same samples (Fig. 1C). We found a total of 756 proteins identified in the samples, and of these 183 showed a decrease of ≥2-fold in unique peptide counts between control and ABT-737 or ABT-199 treatment. Reactome analysis revealed that the decreased proteins were largely constitutive EV factors. Peptide counts confirmed the depletion of insulin in EVs from the senolytic treated islets and depletion of known SASP factor Flnb in the EVs, validating the efficacy of the senolytic treatments, whereas again, Actin was no difference between the samples (Fig. 1D). These data suggest that senescent β-cells export pro-insulin in EVs. Although the human islet senescence model using DNA damage as a trigger for senescence recapitulates several phenotypes observed in β-cells of T1D donors, islets are a mixed cell population, and it remains unclear what the contribution of β cells is to the phenotypes observed.
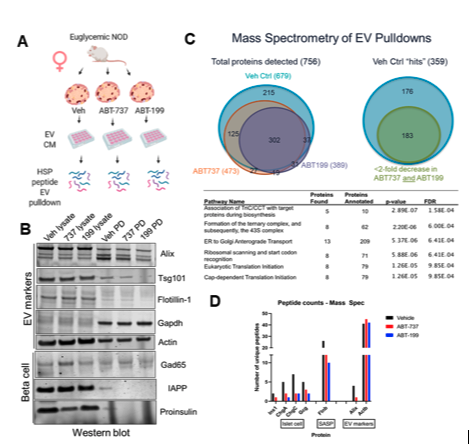
Figure 1. NOD mouse senescent β cell removal decreases sEV release of autoantigen proinsulin. (A) EV pulldown by Heat-shock peptide(Ghosh et al. 2014 PLOS ONE) from isolated islets from NOD mice treated senolytic compounds ABT-737, ABT-199 or Vehicle control (DMSO) for 48 h and western blot for sEV and β cell markers. (B) Western blot on EV pulldowns and islet lysates from indicated samples. (C) Mass spectrometry of samples from (A) showing Venn diagrams of identified hit proteins (>2 unique peptides in Control) and Reactome analysis terms for proteins reduced by >2-fold in senolytic vs control sample. (C) Unique peptide counts for indicated proteins in each sample reveals loss of SASP factor Flnb, EV marker Alix, and islet proteins, while Actin remained unchanged.
6-Month Project Update
The project has two aims, both of which have started. The first aim is to identify the specific antigen-presenting molecules carried by EVs derived from the senescent beta cells by using mass spectrometry. To accomplish this, EVs have been isolated from senescent beta cells and healthy beta cells. The analysis of specific peptides and proteins that carried by the EVs has been initiated. We found a total of 756 proteins identified in the samples. The next step is to perform western blot and flow cytometer to validate the specific peptide or proteins in the EVs from senescent beta cells.
In addition, we also established a co-cultivation system to explore the effects of EVs on immune regulation. We observed that when splenic T cells are co-cultivated with senescent cells, splenic T cells showed an increased expression of activation maker and cytokine release, as compared with the co-cultivation with non-senescent cells. This observation suggests that EVs that derived from senescent cells impact the activation of splenic T cells. Our next step is to test whether the specific proteins that carried by the EVs from senescent cells play a role on the activation of splenic T cells.
Project Description
Type 1 diabetes (T1D) is a chronic inflammatory disease, which affects nearly 1.6 million Americans. In the classical view of T1D, pancreatic beta cells are mistakenly destroyed by immune systems. While the classical view of T1D still holds, further evidence shows that beta cells play a vital role in T1D development and are not simply passively destroyed. Our laboratory illustrated that a specific type of beta cells, aging beta cells, accumulate with the progression of T1D in both rodents and humans.
Furthermore, the elimination of these beta cells halted the immune-mediated beta-cell destruction and prevents T1D in the mouse model. Of note, aging cells can normally be recognized and removed by the immune system. In the T1D model, however, patrolling immune cells fail to recognize and eliminate this type of beta-cell, causing them to accumulate and spread in the pancreas.
The failure of immune cells in T1D development raises a couple of questions: how do the aging β-cells evade immune removal in T1D? Do these cells play a role in wider immune regulation with T1D?
Aging, or senescent, cell communication depends on the release of senescence-associated secretory phenotypes (SASPs), which affect the function of bystander cells. Extracellular vesicles (EVs) are a group of SASPs which carry and transfer various proteins, RNA, and lipids that derived from parental cells. EVs function as carriers of signals that regulate the immune response in T1D.
In this context, senescent beta cells might regulate the immune response via the release of EVs. The goal of this project is to determine the role of EVs that are secreted from senescent beta cells in the ultimate development of T1D. These findings will then lead to a comprehensive understanding of why these cells accumulate with T1D development.
To determine the role of EVs secreted from senescent beta cells, we will first identify the specific antigen-presenting molecules carried by EVs derived from the senescent beta cells, with comparison to those carried by normal EVs. Second, we will examine whether EVs that derived from senescent beta cells can be taken up by the typical antigen-presenting cells. Third, we will evaluate whether EVs that derived from senescent beta cells can affect native T cell activation, which ultimately plays a role in pancreatic beta cells destruction.
In addition to the mouse model, we will use pancreatic beta cells from human donors. Therefore, our findings are more relative to T1D in humans and could be more applicable for therapeutic purposes.
To view Dr. Wang’s video, click HERE.





