

In honor of Matt Polakow, the Tarson Family funded this project in its entirety.
Final Project Update
Since patients with Type 1 diabetes actively lose their active and healthy beta-cell mass in the pancreatic islets, conserving and/or restoring the healthy beta-cell population is critical for preventing disease progression. Previous findings from our lab have established the role of senescent beta cell mass as active drivers of Type 1 Diabetes, which consummates T1D by making active beta cells senescent. This has led us to actively focus on the strategies to selectively target these cells.
Our lab has identified a unique subset of the immune population that actively and preferentially targets this SASP secreting cell mass. We have successfully established through our current work that activation of these specific types of immune cells with a small molecule not only decreases the senescent cell burden from the islets but also improves the metabolic profile.
In the preclinical settings, we are using an exogenous antigen to stimulate these cells, and we observe a significant improvement in ITT and GTT profile of the NOD mice, which correlates with the decrease in percentage of senescent cells in the islets. A brief pathway on how beta cells are rescued is shown in Figure 1.
Click HERE to view the full project update as well as the figure mentioned above.
6-Month Project Update
Since patients with Type 1 diabetes actively lose their active and healthy beta-cell mass in the pancreatic islets, conserving and/or restoring the healthy beta-cell population is critical for preventing disease progression. Previous findings from our lab have established the role of senescent beta cell mass as active drivers of Type 1 Diabetes, which consummates T1D by making active beta cells senescent. This has lead us to focus on the strategies to target these cells actively selectively.
Our lab has identified a unique subset of the immune population that actively and preferentially targets this SASP secreting cell mass. We have successfully established through our current work that activation of these specific types of immune cells with a small molecule not only decreases the senescent cell burden from the islets but also improves the metabolic profile.
In the pre-clinical settings, we are using an exogenous antigen to stimulate these cells, and we observe a significant improvement in ITT and GTT profile of the NOD mice, which correlates with the decrease in percentage of senescent cells in the islets. A brief pathway on how beta cells are rescued is shown in Figure 1.
To this date, we have minimal knowledge about the endogenous ligands for this particular subset of immune cells and as to what regulates their turnover. It is exciting and very promising to harness this endogenous immune surveillance as a therapeutic strategy, as it not only provides new treatment alternative for Type 1 Diabetes but also provides a strong supportive treatment option to deal with various other disorders that have associated auto-immune character including Rheumatoid arthritis, obesity, and pulmonary fibrosis.
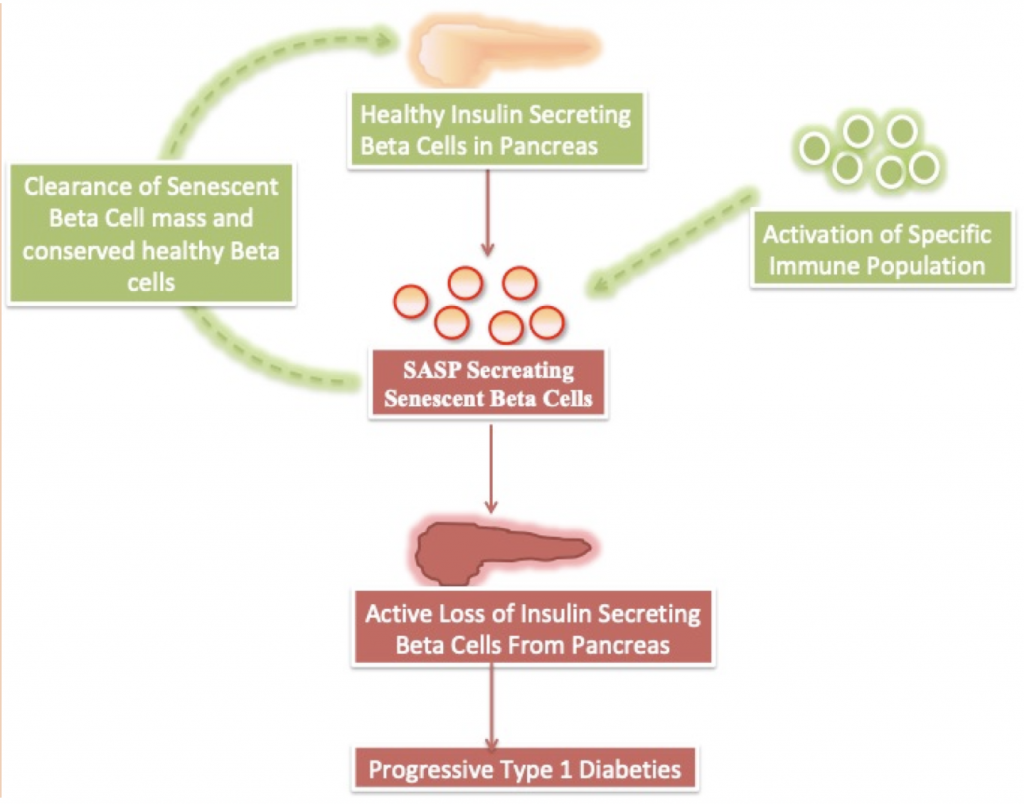
Figure 1: Activation of specific immune subset mitigates Type 1 Diabetes Progression
PROJECT DESCRIPTION
Type 1 Diabetes (T1D) is a chronic disorder in which patients actively lose the insulin-secreting beta cells in the pancreatic islets. Despite extensive research to cure T1D that ranges from insulin treatment to transplanting healthy beta cells, there is no unified hypothesis for the mechanisms that initiate and sustain the loss of beta cells.
Recent research from our lab clearly shows the role of dying beta cells (senescence) in the progression of T1D. Preliminary data also suggests certain substances can increase the production of compensating chemicals to substantially reduce the death of beta cells. These agents also helped in reversing the progression of T1D in mice.
Two major issues arise from these findings: 1) what triggers the death of beta cells in the pancreas of T1D mice, and 2) how to translate this information to humans.
Current work from our lab strongly suggests that a deficient DNA repair machinery results in the accumulation of structured problems in mice beta cells, Double Strand Breaks (DSB), that causes beta-cell death. We propose to develop a unique humanized mouse model that will allow us to test the clearance of these dying human beta cells using a unique humanized mouse model.
Research Project and Approach
To test the above hypothesis, our specific objectives in the proposed study are:
1) Determine how beta cells die as a result of DSB which may be the cause of deficient DNA repair machinery; and
2) Use a humanized mouse model to test the targeted clearance of senescent human beta cells in vivo.
The answers to the above questions will not only provide substantial advancement in our understanding of T1D but may also support the development of a new approach for preventing, and possibly curing the disease. The exact triggers and precise mechanism which causes beta cells to die during the onset of T1D are still unclear. However, it is known that persistent DSB is capable of inducing beta cell death. Current work from our lab strongly suggests the presence of DSB in infiltrated islets of humanized T1D mice. Experimental data from other labs evidences that the deletion of an important enzyme of DSB machinery causes beta cells, in the humanized mouse, to completely resist chemically induced T1D. We will test this hypothesis by removing this enzyme to determine if this may prevent the onset and persistence of T1D.










